What is SDFA
SDFA is an efficient analysis tool designed for large-scale structural variation (SV) analysis. It is based on a new SV storage format and constructs a supporting toolset. Specifically, it first designs a standardized decomposition format (SDF) for SV, which efficiently represents, stores, and retrieves any type of SV data by decomposing SV. Based on the SDF file, SDFA designs or optimizes existing SV analysis algorithms considering the performance in large-scale samples.
What is SDF
The full name of SDF is Standardized Decomposition Format (SDF). It is a novel file format based on columnar blocks, which is used for splitting, storing, and compressing SV data. Its file structure is as follows:
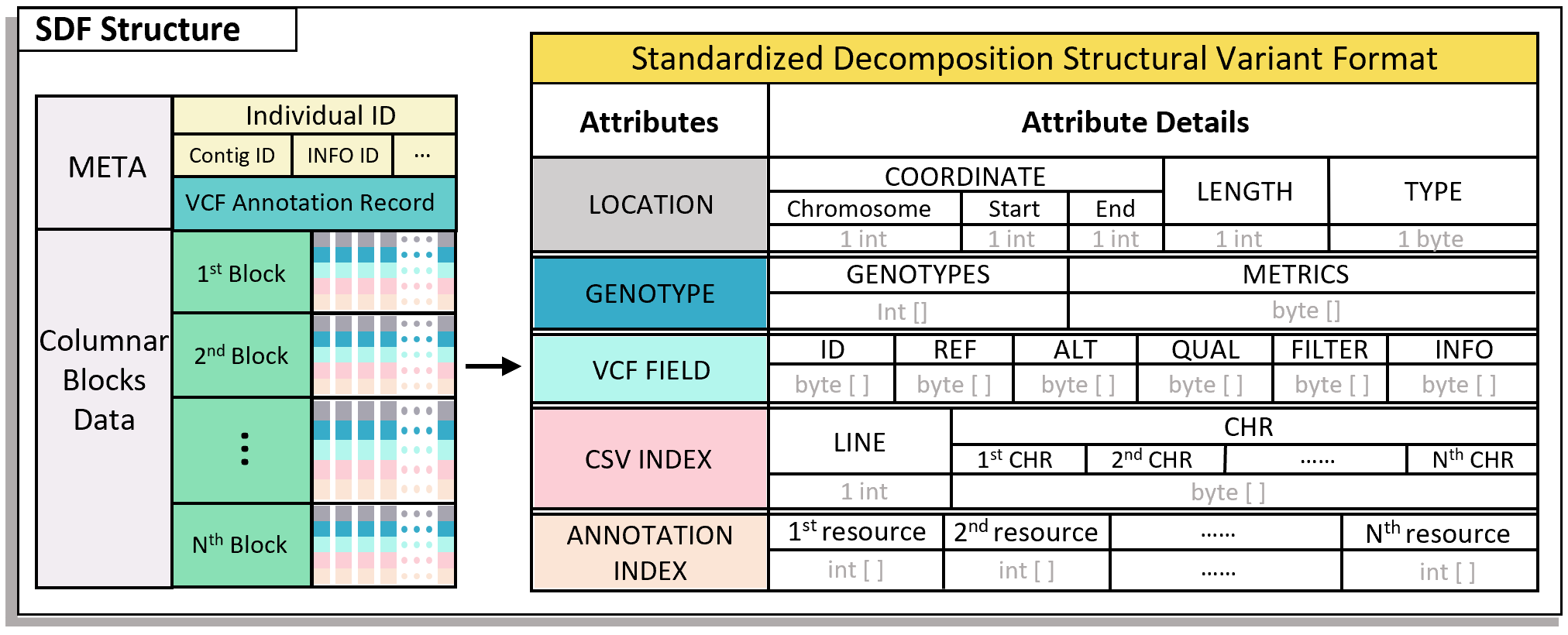
The SDF file is a core component of the SDFA tool and serves as the basic processing file format for downstream SV analysis in SDFA.
Why use SDFA
Compared with existing tools, SDFA has the following advantages:
SDFA provides a systematic solution to fundamentally solve the problems of large-scale SV basic analysis.
It can efficiently handle complex SV types, such as nested SVs, while other tools often fail to correctly parse these complex SVs.
SDFA significantly outperforms existing tools in terms of speed and efficiency, especially on large - scale datasets.
- SDFA can collaborate with tools such as Plink to conduct SV-based GWAS research and explore SV at the population level.
Functions and Outstanding features of SDFA
Efficient SV data storage and retrieval: Achieved through the SDF format.
Consistent and robust SV merging algorithm: Capable of handling large-scale sample data.
Fast and memory-efficient SV annotation: Using the indexed sliding window algorithm.
Novel and precise gene feature annotation: Using the Numerical Annotation of Gene Features (NAGF) method.
Excellent performance: At least 17.64 times faster in SV merging speed and at least 120.93 times faster in annotation speed.
Ability to parse and annotate complex SVs: The only tool that can correctly handle nested complex SVs.
High scalability: Successfully processed 895,054 SVs from 150,119 individuals in the UK Biobank dataset, while other methods failed.
Parallel processing capability: Can utilize multi-threading to improve processing speed.
Flexible customization features: Such as user-defined filtering conditions and annotation resources.