
| Home |
| Download |
| Register |
|
Short Tutorials Mendelian Disease Double Hit Gene Genetic Association Mutation burden Cancer driver |
| Online Manual |
| Simple Demo |
| Miaoxin Li |
Links of MX Li's tools:
KGGSeq is a software platform constituted of Bioinformatics and statistical genetics functions making use of valuable biologic resources and knowledge for sequencing-based genetic mapping of variants/genes responsible for human diseases/traits. Simply, KGGSeq is like a fishing rod facilitating geneticists to fish the genetic determinants of human diseases/traits in the big sea of DNA sequences. Compared with other genetic tools like plink/seq, KGGSeq paid more attention downstream analysis of genetic mapping. Currently, a comprehensive and efficient framework was newly implemented on KGGSeq to filter and prioritize genetic variants from whole exome sequencing data. |
 |
Important new features of KGGSeq v1.0+ you may be interested in: 1. Improved capacity to process whole genome sequencing data of large sample IN PARALLEL with REASONABLE AMOUNT of memory, say, < 10GB; |
|
| Comments and suggestions are welcome, please e-mail limx54@163.com | |
| References: | |
|
1. Li MX, Gui HS, Kwan JS, Bao SY, Sham PC. A comprehensive framework for prioritizing variants in exome sequencing studies of Mendelian diseases.Nucleic Acids Res. 2012 Apr;40(7):e53. PubMed NAR‹For the entire filtration, annotation and prioritization framework› 2. Li M, Li J, Li MJ, Pan Z, Hsu JS, Liu DJ, Zhan X, Wang J, Song Y, Sham PC. Robust and rapid algorithms facilitate large-scale whole genome sequencing downstream analysis in an integrative framework. Nucleic Acids Res. 2017 Jan 23. pii: gkx019. doi: 10.1093/nar/gkx019 PubMed‹For the entire filtration, annotation and prioritization framework with whole genome sequencing data› 3. Li MX, Kwan JS, Bao SY, Yang W, Ho SL, Song YQ, Sham PC. Predicting Mendelian disease-causing non-synonymous single nucleotide variants in exome sequencing studies. PLoS Genet. 2013 Jan;9(1):e1003143. PubMed PLoS Genet‹For Mendelian pathogenic prediction of non-synonymous variants by logistic regression› 4. Hsu JS, Kwan JS, Pan Z, Garcia-Barcelo MM, Sham PC, Li M. Inheritance Modes Specific Pathogenicity Prioritization (ISPP) for Human Protein Coding Genes. Bioinformatics. 2016 Oct 15;32(20):3065-3071.‹For causal prediction at gene levels › 5. Li MJ, Pan Z, Liu Z, Wu J, Wang P, Zhu Y, Xu F, Xia Z, Sham PC, Kocher JP, Li M, Liu JS, Wang J. Predicting regulatory variants with composite statistic. Bioinformatics. 2016 Sep 15;32(18):2729-36.‹For regulatory potential prediction at non-coding variants › 6. Li MJ, Li M, Liu Z, Yan B, Pan Z, Huang D, Liang Q, Ying D, Xu F, Yao H, Wang P, Kocher JA, Xia Z, Sham PC, Liu JS, Wang J. cepip: context-dependent epigenomic weighting for prioritization of regulatory variants and disease-associated genes, Genome Biology (2017) 18:52 ‹For tissue- or cell type specific regulatory potential prediction at non-coding variants › 7. Jiang et al. WITER: a powerful method for estimation of cancer-driver genes using a weighted iterative regression modelling background mutation counts. Nucleic Acids Res. 2019 Sep 19;47(16):e96. doi: 10.1093/nar/gkz566. PubMed ‹For mutation burden test at cancer-driver genes› 8. Jiang et al. Deviation from baseline mutation burden provides powerful and robust rare-variants association test for complex diseases. Nucleic Acids Res. 2021 Dec 20. doi: 10.1093/nar/gkab1234. PubMed ‹For mutation burden test at genes with rare variants; RUNNER› |
|
|
|
|
|
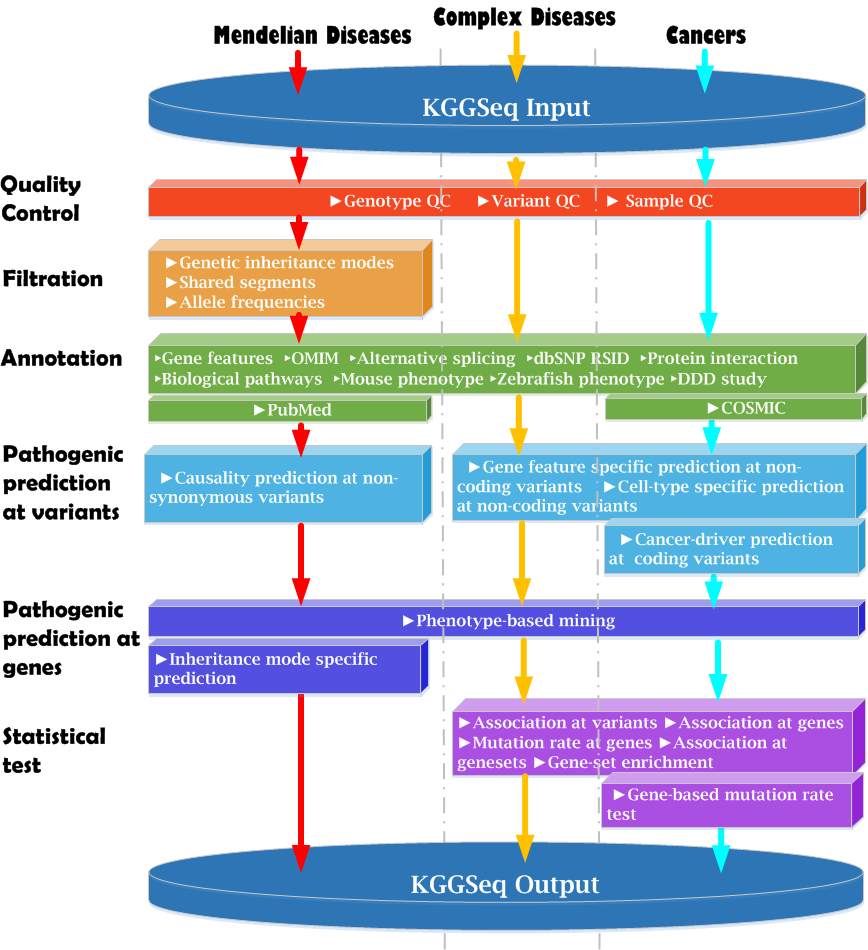
The pipeline of KGGSeq V1.0+ : |
|
|
|