压缩基因型
使用如下指令对基因组 VCF 文件进行压缩:
build <input(s)> -o <output> [options]
- GBC 可以为符合 VCF 文件规范 的文件进行压缩,GBC 的所有操作都是假定文件格式是符合此规范的;
- 输入文件可以是单个或多个 .vcf 文件或 .vcf.gz 文件,也可以是包含这些文件的文件夹路径。当路径为文件夹路径时,GBC 会筛选出该文件夹 (及其子文件夹) 中所有的 vcf 文件或 .vcf.gz 文件进行压缩。请注意,GBC 仅根据文件的扩展名判断文件类型,因此正确的文件扩展名才能够进行压缩;
- 为了便于统一全局的符号,GBC 当前仅支持对于人类基因组的压缩,CHROM当前只支持 1-22, X, Y, MT 以及带有 chr 前缀的 1-22, X, Y, MT;对于其他物种,需要先构建染色体标签文件;
- GBC 对多个文件进行合并压缩时,要求这些文件具有相同的样本序列 (样本顺序可以不一致)。若一个文件的样本序列是其他文件的子序列,它也可以被正确压缩,缺失的样本基因型将被替换为 .|.;
- 当输入的文件在坐标上无序时,GBC 也能正确地压缩,并产生标记为
unordered的 GTB 文件。通常我们建议用户进一步使用sort <input> -o <output> [options]指令对该文件进行排序,否则它将在某些功能上不可用 (例如: 计算 LD 系数) 或影响其他操作的性能 (例如: 合并 VCF 文件)。
程序参数
语法: build <input(s)> -o <output> [options]
参数:
--contig 指定染色体标签文件.
默认值: /contig/human/hg38.p13
格式: --contig <file> (Exists,File,Inner)
*--output,-o 设置输出文件名.
格式: --output <file>
--threads,-t 设置并行压缩线程数.
默认值: 4
格式: --threads <int> (>= 1)
--yes,-y 覆盖输出文件.
GTB 存档参数:
--phased,-p 设置基因型数据为 phased.
--biallelic 将多个多等位基因位点分裂为多个二等位基因位点.
--simply 删除 ALT 中等位基因计数值为 0 的标签.
--blockSizeType,-bs 设置每个压缩块的最大位点数, 根据 2^(7+x) 换算得到真实的块大小值.
默认值: -1 (即根据样本量自动设置)
格式: --blockSizeType <int> (-1 ~ 7)
--no-reordering,-nr 禁用 Approximate Minimum Discrepancy Ordering (AMDO) 算法.
--windowSize,-ws 设置 AMDO 算法的采样窗口大小.
默认值: 24
格式: --windowSize <int> (1 ~ 131072)
--compressor,-c 设置基压缩器.
默认值: ZSTD
格式: --compressor <string> ([ZSTD/LZMA/GZIP] or
[0/1/2] (ignoreCase))
--level,-l 基压缩器的压缩级别. (ZSTD: 0~22, 默认为 3; LZMA: 0~9,
默认为 3; GZIP: 0~9, 默认为 5)
默认值: -1
格式: --level <int> (-1 ~ 31)
--readyParas,-rp 从外部 GTB 文件中导入模版参数 (-p, -bs, -c, -l).
格式: --readyParas <file> (Exists,File)
--seq-ac 移除等位基因计数不在 [minAc, maxAc] 范围点的位点.
格式: --seq-ac <int>-<int> (>= 0)
--seq-af 移除等位基因频率不在 [minAf, maxAf] 范围点的位点.
格式: --seq-af <double>-<double> (0.0 ~ 1.0)
--seq-an 移除有效等位基因个数不在 [minAn, maxAn] 范围点的位点.
格式: --seq-an <int>-<int> (>= 0)
--max-allele 移除等位基因种类超过指定值的位点.
默认值: 15
格式: --max-allele <int> (2 ~ 15)
质量控制参数:
--no-qc 禁用所有的质量控制方法.
--gty-gq 样本基因型 GQ 分数值小于指定值时, 该基因型将被替换为 '.|.'.
默认值: 20
格式: --gty-gq <int> (>= 0)
--gty-dp 样本基因型 DP 分数值小于指定值时, 该基因型将被替换为 '.|.'.
默认值: 4
格式: --gty-dp <int> (>= 0)
--seq-qual 移除 Phred Quality 分数值小于指定值的位点.
默认值: 30
格式: --seq-qual <int> (>= 0)
--seq-dp 移除 DP 分数值小于指定值的位点.
默认值: 0
格式: --seq-dp <int> (>= 0)
--seq-mq 移除 MQ 分数值小于指定值的位点.
默认值: 20
格式: --seq-mq <int> (>= 0)
程序实例
使用 GBC 压缩示例文件 ./example/assoc.hg19.vcf.gz,并设置以下参数:
- 基因型设置为 phased;
- 压缩器压缩级别设置为 16;
- 将多等位基因位点分裂为二等位基因位点;
- 保留 MAF (次级等位基因频率) > 0.01 的位点;
- 若输出文件已存在,则覆盖该文件。
完成该任务的指令如下:
# Linux 或 MacOS
docker run -v `pwd`:`pwd` -w `pwd` --rm -it -m 4g gbc \
build ./example/assoc.hg19.vcf.gz -o ./example/assoc.hg19.gtb -p -l 16 --biallelic --seq-af 0.01-0.99 -y
# Windows
docker run -v %cd%:%cd% -w %cd% --rm -it -m 4g gbc build ./example/assoc.hg19.vcf.gz -o ./example/assoc.hg19.gtb -p -l 16 --biallelic --seq-af 0.01-0.99 -y
算法介绍
GBC 压缩算法的流程分为分块、字节编码、最大化字节编码、位点的 AMDO 排序、使用基压缩器压缩数据流、储存到 GTB 文件。该算法对于样本和位点的数量具有线性时间复杂度,内存使用量小 (小于4GB) ,而且它提供了迄今为止几乎最快的压缩速度和最具竞争力的压缩比。
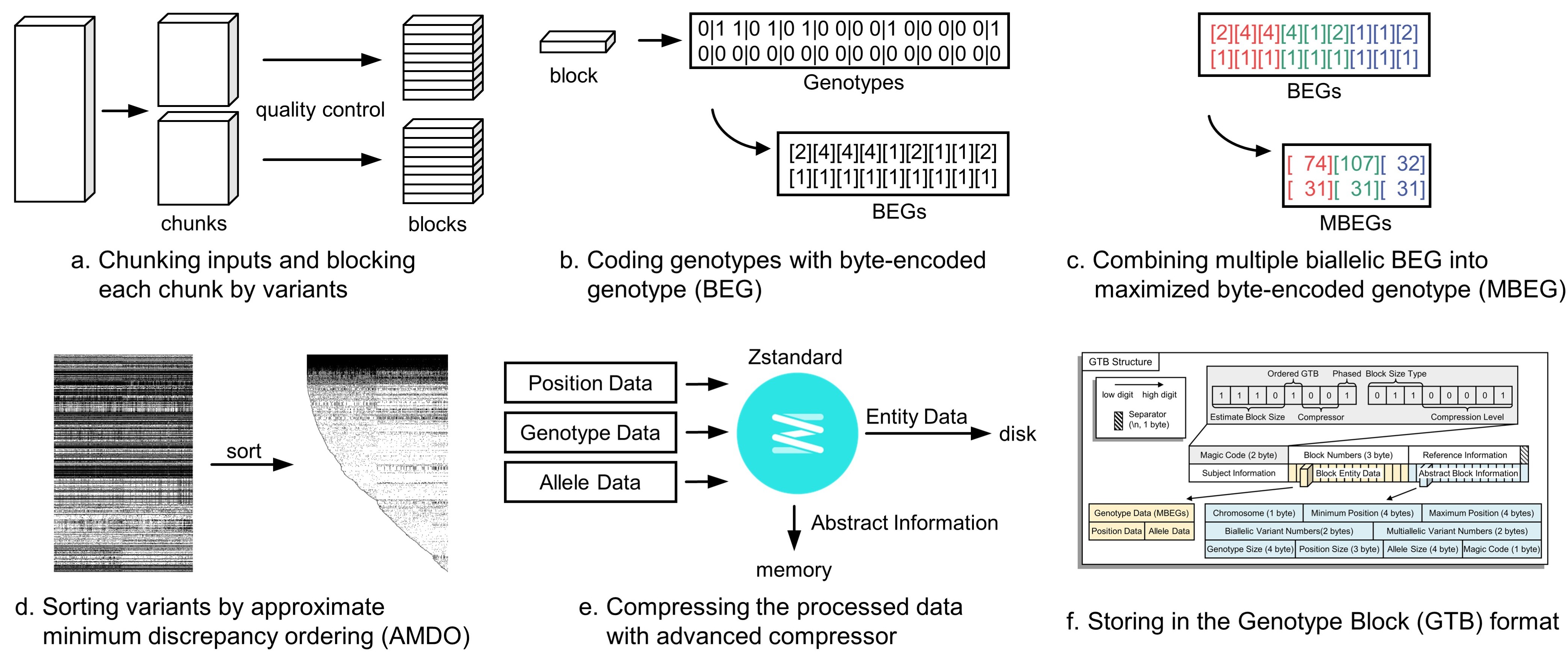
文件输入
多个文件输入时,GBC 将具有最多样本的文件作为母文件,其他的文件 (子文件) 的样本顺序则与母文件对齐。具体对齐策略如下:
- 当子文件的样本不包含在母文件中时,程序报错;
- 当子文件的样本顺序与母文件的样本顺序不一致时,子文件样本的基因型顺序将按照母文件的样本顺序进行重排;
- 当母文件的样本不包含在子文件中时,缺失的样本将以
.|.或./.代替。
[!NOTE|label:适用情形]
不少人类基因组项目 (如 1000GP3、SG10K) 都会将基因型按照不同染色体编号进行存放 (即 chr1.gz, chr2.gz, ...)。此外,在 Y 染色体文件中通常不包含雌性个体,造成 Y 染色体文件的样本数通常小于常染色体或 X 染色体。
这种策略可以将基因组的多个散布的文件合并压缩,也可以将人类基因组中的性染色体文件与常染色体文件合并压缩。
初始化
为控制每个线程的最大内存用量,当总样本量 被确定时,每个 GTB 块的最大位点数 也就被确定。通常情况下,样本量范围对应的块大小参数 (或块位点个数) 关系表如下:
| 参数 | 参数 | ||||
|---|---|---|---|---|---|
| -bs 7 | 16384 | -bs 3 | 1024 | ||
| -bs 6 | 8192 | -bs 2 | 512 | ||
| -bs 5 | 4096 | -bs 1 | 256 | ||
| -bs 4 | 2048 | -bs 0 | 64 |
其中,。
文件分块以并行化
输入的文件依次进行压缩。在每个文件压缩时,按照并行线程数 被近似地均等分成 块,每个线程处理 1 个块。当输入文件为 BGZIP 压缩的 VCF 格式时,GBC 会检查块边界处的信息、调整指针,确保块中的位点都是完整的。
位点和基因型水平质控
线程在处理单个块时,按照行读取位点,并解析非基因型字段。通过非基因型字段预先进行初步的位点水平质量控制 (例如: 根据 Phred Quality Score、MQ 进行质控),当位点不满足质控要求时,线程继续读取下一个位点。
满足位点水平质控的位点则对其基因型进行质控。当位点 FORMAT 的值为 GT 时,不进行基因型水平的质控;若该值还包含了其他的关键字段,则对于每一个读取的基因型将进行相应的质量控制 (例如: 根据 DP、GQ 进行质控)。当基因型不满足质控要求时,该基因型被 .|. 替代。
编码基因型为字节码 (BEG)
满足基因型水平质控的基因型进行字节编码。对于给定的位点 , 非缺失基因型 编码为:
即:。对于特殊情形,约定如下:
- 缺失基因型 编码为 0;
- 非分相 (unphased) 基因型 被转换为 后按照上式编码;
- 单倍型 被转换为 后按照上式编码。
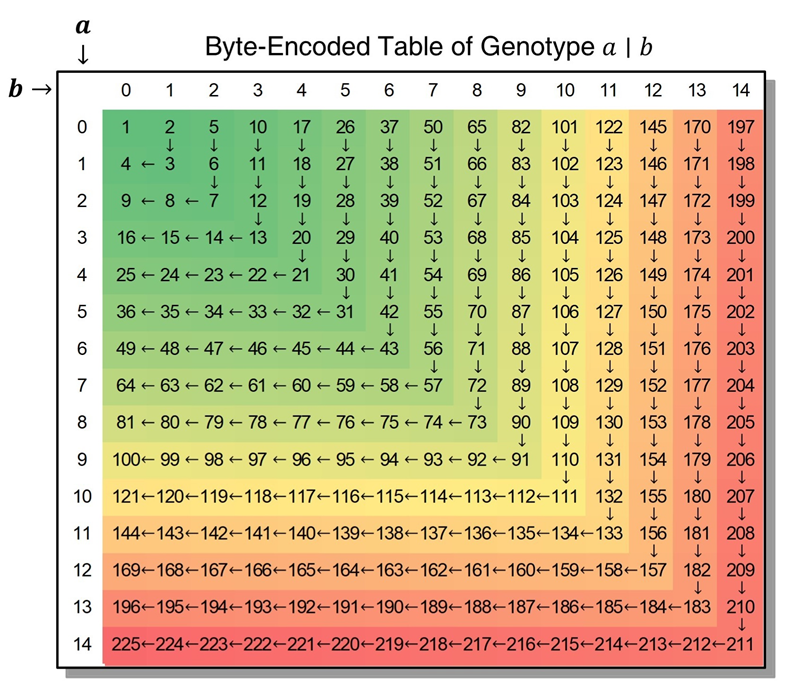
将位点写入 GTBWriter 缓冲区
当位点的基因型都被编码完成时,该位点被传输到 GTBWriter 缓冲区,以进入后续的压缩环节。在这一步,位点被执行更多的过滤或转换操作:
- 当启动了 “--simply” 时:多等位基因位点将移除等位基因计数为 0 的 ALT 标签,基因型则根据新的等位基因标签重新编码为字节值;
- 当启动了 “--biallelic” 时:多等位基因位点被拆分为多个二等位基因位点;
- 当启动了位点水平质控时:该位点进行位点水平质量控制,当位点不满足质控要求时,该位点将被跳过;
当 GTBWriter 缓冲区大小没有达到 GTB 块的最大位点数 或该分块文件没有读取结束时,重复进行以上的位点读取、质控、编码、写入缓冲区的操作;否则,进行以下的压缩环节。
基因型阵列进行特征重排列 (AMDO)
缓冲区中的位点按照如下方式进行重排列,以提升压缩比:
Step 1: 样本 在位点 上的基因型的 "0" 等位基因个数, 记为 ;
Step 2: 生成降采样序列 , 其中: 式中 为总样本数, 为连续采样基因型的个数;
Step 3: 根据降采样序列, 二等位基因 (biallelic) 位点进行正向字典序排序。多等位基因 (multiallelic) 位点进行反向字典序排序。位点 和位点 的顺序被定义为 和 的字典序,即:
- 如果 , 使得 , 则 ;
- 如果 , 使得 , 则 ;
- 如果 , 使得 , 则 。
最终,获得位点的新索引 。
将基因型字节码 (BEG) 转为最大化字节码 (MBEG)
对于二等位基因 (biallelic) 位点, 连续的 3~4 个基因型按照以下方法组合为单个字节:
当位点基因型的个数不构成 3 或 4 的倍数时, 末尾处的"缺失基因型"被设置为上一个非缺失值.
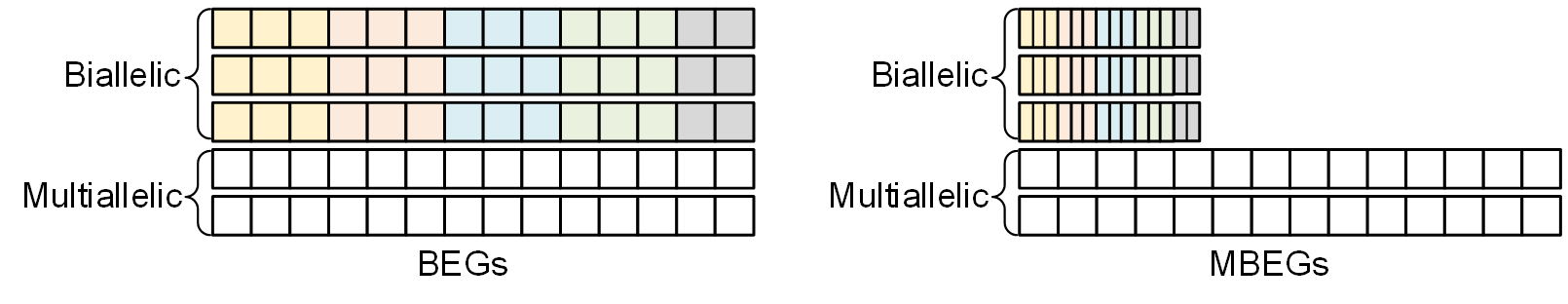
合并数据流并压缩
每个区块中的所有排序的 MBEG 和 BEG 代码都由先进的压缩器进一步压缩。流行的压缩算法(如 ZSTD、LZMA、GZIP 等)在基因型上可以达到 100 倍以上的压缩率。我们默认选择了ZSTD,因为在广泛使用的压缩算法中,它提供了最快的压解速度。
具体而言,每个位点的基因型编码都直接串联成一个字节数组 。接下来,每个位点的位置被转换为 4 个字节,然后块内所有位点的位置被串联到一个字节数组 。最后,所有变体的等位基因被串联到另一个带有 / 分隔符的字节数组。然后,这些串联的数据 、 和 被基压缩器压缩,分别产生 、 和 ,这三个部分组成了块的压缩数据。块数据会即时写入线程相应的输出文件。
此外,根据压缩情况,每个块对应产生一个摘要信息:染色体编号(1个字节)、最小和最大位置(分别为4个字节)、二等位基因位点的数量(2个字节)、多等位基因位点的数量(2个字节)、 的长度(4个字节)、 的长度(3个字节)、 的长度(4个字节)和块的校验编码(1个字节)。摘要信息被写入线程的内存队列中。
GBC 可以与不同的压缩算法相结合。ZSTD、LZMA、GZIP 算法已被嵌入,我们还保留了 1 种类型的压缩器,供开发者在未来扩展。
完成压缩
当一个线程完成压缩时,块摘要信息被拼接到线程相应的输出文件,文件指针挪动到文件头部,并修改块头信息。
当所有的线程都完成压缩时,GBC 主程序调用 Concat 方法将每个线程产生的 GTB 文件拼接成一个整体的 GTB 文件。