Split GTB
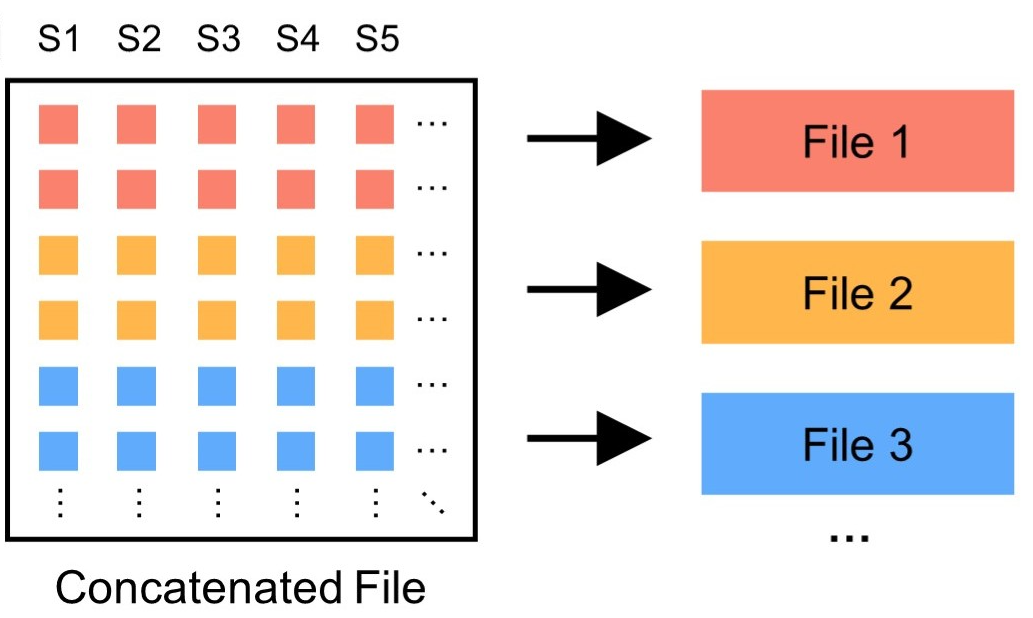
Splitting a GTB file into multiple subfiles is a common requirement when the size of a single GTB file is quite large. Because this facilitates program debugging and file transfer. A typical example is the whole exome genotype of UKBB, and the genotypes on chromosome 1 is split into 97 subfiles. Use the following command to split a GTB file into multiple smaller subfiles:
java -jar gbc.jar split <input> [output] [options]
The split subfile can be re-joined by the GTBConcat.
[!NOTE|label:Example|style:callout]
Use GBC to split the example file
https://pmglab.top/gbc/download/rare.disease.hg19.gtbinto multiple subfiles according to the chromosome tag of the variants.# Run directly in the terminal java -jar gbc.jar split https://pmglab.top/gbc/download/rare.disease.hg19.gtb # Run it using docker docker run -v `pwd`:`pwd` -w `pwd` --rm -it -m 4g gbc \ split https://pmglab.top/gbc/download/rare.disease.hg19.gtb
Program Options
Usage: split <input> [outputDir] [options]
Java-API: edu.sysu.pmglab.gbc.toolkit.GTBSplitter
About: Split a single *.gtb file into multiple sub-files (e.g., split by chromosome or variant index).
Options:
--chromosome Specify the chromosome tags file. e.g., identify 'X, chrX,
CHRX, ChrX' as '(int) 22' chromosome.
format: --chromosome <file>
--threads,-t Set the number of threads.
default: 4
format: --threads <int>
--by Split input file by chromosome-level or variant-level into
multiple sub-files, which can be rejoined by the concat mode.
default: chromosome
format: '--by chromosome [tag],[tag],...' or '--by variant
[int]'
Subset Selection Options:
--pos,-p Retrieve the variants by the specified coordinates of
variant.
format: --pos <chr>:<pos>,<pos>,... ... (>= 1)
--pos-range,-pr Retrieve the variants by the specified coordinate
intervals of variant.
format: --pos-range <chr>:<minPos>-<maxPos>,... (>= 1)
--index-range,-ir Retrieve the variants by the line-index of variant.
format: --index-range <minIndex>-<maxIndex> (>= 0)
--allele-num Exclude variants with the alternative allele number per
variant out of the range [minAlleleNum, maxAlleleNum].
format: --allele-num <minAlleleNum>-<maxAlleleNum> (0 ~
255)
--seq-ac Exclude variants with the alternate allele count (AC) per
variant out of the range [minAc, maxAc].
format: --seq-ac <minAc>-<maxAc> (>= 0)
--seq-af Exclude variants with the alternate allele frequency (AF)
per variant out of the range [minAf, maxAf].
format: --seq-af <minAf>-<maxAf> (0.0 ~ 1.0)
--seq-an Exclude variants with the non-missing allele number (AN)
per variant out of the range [minAn, maxAn].
format: --seq-an <minAn>-<maxAn> (>= 0)
--field-condition Extract variants by the values of the specified
supplementary fields. For comparable fields, the
'condition' format is 'minValue-maxValue'; for other
formats, the 'condition' is multiple optional values
separated by ','.
format: --field-condition <field>=<condition>
<field>=<condition> ...
Edit Meta Options:
--add-meta Add the specified metas to the output file.
format: --add-meta <key>=<value> <key>=<value> ...
--rm-meta Remove all meta information.
API Toolkit
The API tool for splitting GTB files is edu.sysu.pmglab.gbc.GTBSplitter. Example usage is as follows:
GTBSplitter.of("https://pmglab.top/gbc/download/rare.disease.hg19.gtb")
.splitByChromosome(null);