Basic Data Objects
Introduction
GBC has designed multiple API objects around the common structure of the human genome, with a data model similar to the VCF file specification. Different with GA4GH Schemas, GBC only requires the mandatory coordinate fields for variant (i.e., CHROM and POS), while other fields use key-value pairs (type of key is Object) to store as variant's property. This approach helps to share genomics data in a standardized way, saving the fixed memory overhead required for non-essential fields, as well as being broadly compatible with different API implementations. The following is a list of the most commonly used APIs:
- edu.sysu.pmglab.gbc.variant.Chromosome: Chromosome object.
- edu.sysu.pmglab.gbc.variant.Strand: Chain Direction enumeration class , with 2 enumeration instances: FWD (forward strand) and REV (reverse strand). The strand direction of the variant is forced to FWD. Variants on the reverse strand should be adjusted to FWD using the chromosome length.
- edu.sysu.pmglab.gbc.variant.PositionType: Position type enumeration class, with 2 enumeration instances: ONE_BASED (1-based) and ZERO_BASED (0-based). The position type of the variant is forced to 1-based
- edu.sysu.pmglab.gbc.variant.RefGenomeVersion: Reference genome version enumeration class, with 4 enumeration instances: hg18, hg19, hg38 and unknown. The non-human reference genome can only be used unknown.
- edu.sysu.pmglab.gbc.variant.Variant: Standard variant object.
- edu.sysu.pmglab.gbc.variant.Variants: Collection object for store multiple variants object with the same coordinates.
- edu.sysu.pmglab.gbc.variant.genotype.Genotype: Genotype object.
- edu.sysu.pmglab.gbc.variant.genotype.IGenotypes: Genotype array object.
The following is an example of creating a variant with multiple optional alleles, genotype array, and user-defined property:
// Create a variant
Variant variant = new Variant("chr1", 100, "AT", "A", "C", "TT");
// Create an unphased genotype array
IGenotypes genotypes = new Genotypes(false, 4);
// Set genotype for each subject
genotypes.setGenotype(0, Genotype.of(0, 1));
genotypes.setGenotype(1, Genotype.of(0, 2));
genotypes.setGenotype(2, Genotype.of(1, 2));
genotypes.setGenotype(3, Genotype.of(2, 3));
// Set the property of the variant
variant.setProperty(IGenotypes.class, genotypes);
variant.setProperty("isExon", false);
Chromosome
Chromosome is designed with a global static class to normalize the same entity pointed by different chromosome names. The following is used to obtain a chromosome object with the name "chr1":
Chromosome chromosome1 = Chromosome.get("chr1");
It is associated to the same chromosome object as the chromosome named CM000663.2 in GenBank:
// return true
chromosome1.equals(Chromosome.get("CM000663.2"))
Multiple chromosome names attributed to the same object can be found here: Chromosome Tag Declaration.
Since the chromosome tags system used by different VCF files, different databases may be dissimilar (e.g. chromosomes in dbSNP are named using NCBI Reference Sequence), it is not possible (or requires complex design) to map to the same entity using regular string format for determination. Here, we recommend that all chromosome objects are mapped to the same entity using Chromosome.get(Object object) .
If you want to set other alternative names for the chromosome object or add chromosomes, refer to the following example:
// Clear the original chromosome tags (optional)
Chromosome.clear();
// Create a new chromosome tag
Chromosome chromosome1 = Chromosome.addChromosome(new Chromosome("1", "chr1", "custom1_chr1", "custom2_chr1"));
// Set chromosome's properties
chromosome1.setProperty("length", 249250621);
// get chromosome's properties
chromosome1.getProperty("length");
Position Type
There are two types of coordinate systems PositionType in genomics: 0-based and 1-based. 0-based coordinate system means counting from 0, while 1-based coordinate system means counting from 1. For a sequence ATGC, the base T has coordinates 1 under 0-based and 2 under 1-based, and for the subsequence TG, the coordinates are [1, 3) under 0-based and [2, 3] under 1-based.
Reference Genome Version
The human reference genome versions are published by the Genome Reference Consorium, and the widely used versions are GRCh37 and GRCh38 (hg19 and hg38 are the corresponding UCSC browser versions). The main differences between the two versions are: 1) hg38 has some more sequencing sites than hg19, resulting in shifted positions throughout the genome; 2) hg38 is a huge improvement over hg19, changing some of the previous sequencing errors and assembly errors.
In general, the GBC does not require to specified the reference genome version RefGenomeVersion. Several functions of GBC (e.g., Build GTB for VCF and Export as GTB Fomat) provide the ability to convert the reference genome version. API method edu.sysu.pmglab.gbc.annotation.impl.liftOver.LiftOver is used to provide reference genome version conversion (called liftover in genomics). LiftOver requires a Chain File as input (or use the edu.sysu.pmglab.gbc.annotation.impl.liftOver.LiftOver.Chain class to construct interval mappings). GBC provides four reference genome version: hg18, hg19, hg38, unknown, and built-in liftover of hg18ToHg19, hg18ToHg38, hg19ToHg38, hg38ToHg19. The commonly used Chain files can be downloaded from UCSC website. For example:
- hg19ToHg38: https://hgdownload.soe.ucsc.edu/goldenPath/hg19/liftOver/hg19ToHg38.over.chain.gz
- hg38ToHg19: https://hgdownload.soe.ucsc.edu/goldenPath/hg38/liftOver/hg38ToHg19.over.chain.gz
LiftOver hg19ToHg38LiftOver;
// Use the built-in hg19ToHg38, which downloads the corresponding LiftOver file from the UCSC website
hg19ToHg38LiftOver = LiftOver.load(RefGenomeVersion.hg19, RefGenomeVersion.hg38);
System.out.println(hg19ToHg38LiftOver.convertCoordinate(Chromosome.get("chr8"), 141310715));
// Use the UCSC file address, which can be either a local file path or an http/https file path
hg19ToHg38LiftOver = LiftOver.load("https://hgdownload.soe.ucsc.edu/goldenPath/hg19/liftOver/hg19ToHg38.over.chain.gz");
System.out.println(hg19ToHg38LiftOver.convertCoordinate(Chromosome.get("chr8"), 141310715));
LiftOver has five API methods for converting coordinates:
- convertCoordinate(Chromosome chromosome, int position): performs liftover on the specified coordinate (1-based) and returns the mapped coordinate on the positive strand.
- It returns null when the source coordinates cannot be mapped to the target reference genome version.
- The return value is an
Entry<Chromosome, Integer>object, use getKey() to get the chromosome and use getValue() to get the position.
- convertCoordinate(Chromosome chromosome, int position, PositionType positionType): performs liftover on the specified coordinate (1-based) and returns the mapped coordinate on the positive strand.
- It returns null when the source coordinates cannot be mapped to the target reference genome version.
- The return value is an
Entry<Chromosome, Integer>object, use getKey() to get the chromosome and use getValue() to get the position.
- convertCoordinate(Variant variant): performs liftover on the specified variant.
- It returns null when the source variant cannot be mapped to the target reference genome version.
- The return value is a Variant object with new coordinates, and when the returned Variant object has the same coordinates as the input Variant object, the input value itself is returned.
- convertInterval(Chromosome chromosome,
Interval<Integer>positions): performs liftover on the specified coordinate (1-based) and returns the mapped coordinate on the positive strand.- When the source start or end coordinate cannot be mapped to the target reference genome version, the length of the interval before and after mapping is inconsistent, and the mapped start and end coordinates are not on the same chromosome, it returns null.
- The return value is an
Entry<Chromosome, Interval<Integer>>object, using getKey() to get the chromosome and getValue() to get the position interval.
- convertInterval(Chromosome chromosome,
Interval<Integer>positions, PositionType positionType): performs liftover on the specified coordinate (1-based) and returns the mapped coordinate on the positive strand.- When the source start or end coordinate cannot be mapped to the target reference genome version, the length of the interval before and after mapping is inconsistent, and the mapped start and end coordinates are not on the same chromosome, it returns null.
- The return value is an
Entry<Chromosome, Interval<Integer>>object, using getKey() to get the chromosome and getValue() to get the position interval.
When you are developing a LiftOver-enabled feature (e.g., variant function annotation), we strongly recommend to set the value of "disable liftover" to LiftOver.EMPTY instead of null. This instance is a static member of LiftOver, which returns all inputs as is.
Variant
Single Variant Object
The single variant object Variant is used to represent variation information on a coordinate, and contains two mandatory and immutable member variables:
- chromosome: chromosome object, not allowed to be null.
- position: base position, positive integer, required to be a 1-based coordinate on a positive chain.
and optional member variables:
- alleles: list of optional alleles, the first optional allele corresponds to the REF in VCF file specification, the rest of the optional alleles correspond to the ALT.
addAlleles(String... alleles)oraddAlleles(ByteCode... alleles): add alleles.alleleOfIndex(int index): get the allele at the specified index (the allele corresponding to index 0 is the reference allele), return null if it exceeds the index range.indexOfAllele(String allele)orindexOfAllele(ByteCode allele): get the index of the specified allele, -1 if the specified allele does not contain.isStandardAllele(String allele)orisStandardAllele(ByteCode allele): determine if the specified allele is a standard allele (i.e. all bases are A, T, C, G)isValidAllele(String allele)orisValidAllele(ByteCode allele): determine if the specified allele is a legal allele (i.e. not,;and invisible characters)getAlleles(): get all optional alleles in this variant.getAlleleNum(): get the number of optional alleles in this variant.clearAlleles(): to clear the optional alleles in this variant.
Moreover, use setProperty(Object key, Object value) to set the variant's property and use getProperty(Object key) to get the property. For example, the standard genotype array object in GBC is obtained using getProperty(IGenotypes.class).
Other than the above variables and methods, variant object also provide a method normalized(), which is used to convert a multiallelic variant to multiple biallelic variants and correct backward redundant bases. The return value when normalized() is performed on a single variant object is a collection variant object, which can be used to conveniently represent the functional annotation of each mutation pairs. The following is an example of normalizing a multiallelic vairant:
#CHROM POS REF ALT S1 S2 S3 S4 isExon
1 100 AT A,C,TT 0/1 0/2 1/2 2/3 false
this variant is standardized as:
#CHROM POS REF ALT S1 S2 S3 S4 isExon
1 100 AT A 0/1 0/0 0/1 0/0 false
1 100 AT C 0/0 0/1 0/1 0/1 false
1 100 A T 0/0 0/0 0/0 0/1 false
Note that in this case the genotype number 0 stands for non-ALT genotype and it is necessary to scan the genotypes of all variants with the same coordinates when determining the final genotype of an individual. The properties of the single variant object are set as the properties of the collection variant object. If you need to set the same key property for a child locus, get the child variant locus with a specific index to set via the get(int index) method (priority: same name child locus attribute > same name main locus attribute).
[!TIP|label:Compatible with variant objects from other API specifications|style:callout]
After setting the necessary chromosome and position, variant objects in the other API specification is used as property of the GBC's variant object:
variant.setProperty(org.cobi.kggsee.entity.Variant.class, kggseeVariant);
Collection Variant Object
The collection variant object Variants is used to represent multiple variants with the same coordinates (chromosome, position). It also contains the mandatory chromosome and position. Use add(Variant variant) and addAll(Iterable variants) to add variants to this object. The get(int index) method is used to get the subvariant in this object with index index, while the size() method is used to get the number of subvariants contained in this object.
As with the single variant object, use setProperty(Object key, Object value) to set the property of the variant and getProperty(Object key) to get the property value. At this point, these properties set on the collection variant object represent properties common to all subvariant object's.
Genotype
The genotype class Genotype is used to standardize the genotypes of haploid and diploid species, and it can represent genotypes containing up to 255 optional alleles (i.e., each genotype code in genotype a|b does not exceed 254).
Genotypes are represented in GBC using a singleton object, which causes all genotypes a|b to be located to genotype objects with the same memory address. Missing genotypes use Genotype.of(-1, -1) or Genotype.MISS_GENOTYPE to obtain the genotype object; diploid genotypes a|b or a/b use Genotype.of(a, b) to obtain the genotype object; haploid genotypes a use Genotype.of(a, a) to obtain the genotype object.
The instance object of this class provides the following methods:
- reverse(): reverses the genotype
a|btob|aand returns the genotype instance pointing tob|a. - toUnPhased(): converts genotype
a|bto the UnPhased form (i.e. left genotype right genotype) and returns the genotype instance. - getLeftGenotype(): get the left genotype code of genotype
a|b, i.e.,a. - getRightGenotype(): get the right genotype code of genotype
a|b, i.e.,b. - isMissingGenotype(): whether the genotype is a missing genotype.
- isHomozygous(): whether the genotype is homozygous.
- isHeterozygous(): whether the genotype is heterozygous.
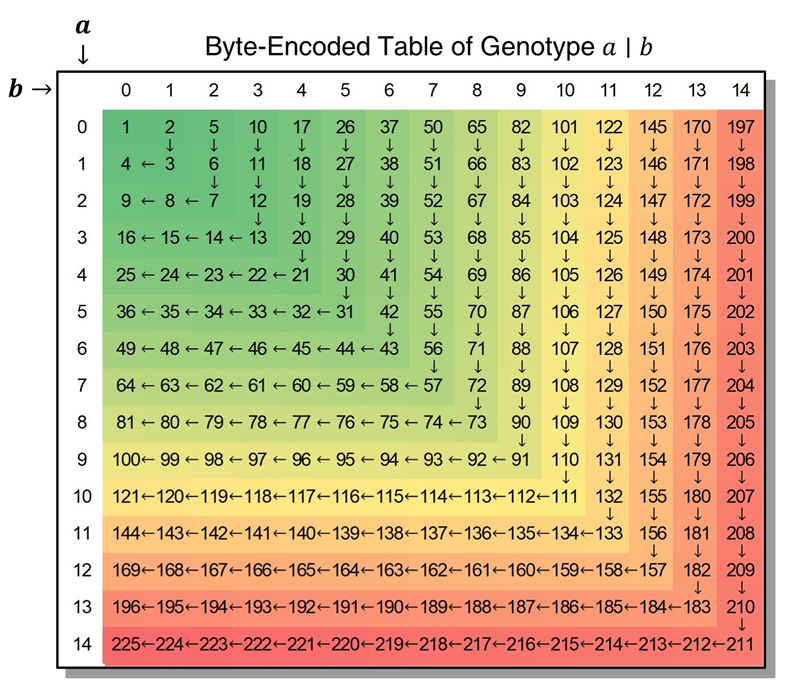
Byte-encodes of Genotype
Each genotype is mapped (hashed) to a unique integer in the range 0-65025. For missing genotypes .|., it is mapped to 0. For a non-missing genotype a|b, it is mapped to
For the complete genotype mapping table, please refer to BEG.xlsx. The genotype can be encoded to single-byte and double-byte forms. When the genotype code on each side does not exceed 14, the genotype is encoded as its single-byte hash value (in this case, the hash value does not exceed 211). When the genotype code exceeds 14 on at least one side, the genotype is encoded as a double-byte hash. For example, the hash value of genotype 20|30 is 921 and its double-byte encoded result is: [-103, 3].
Genotype Array
Genotype array classes IGenotypes are used to store genotypes for multiple subjects on the same coordinate. When a variant contains subject genotypes, these genotypes (as genotype array) are stored on the IGenotype.class property of the variant object. The genotype sequence includes the following common methods:
- getGenotype(int index): get the genotype at the specified index.
- getLeftGenotype(int index): get the left genotype of the specified index.
- getRightGenotype(int index): get the right genotype of the specified index.
- setGenotypes(int index, Genotype genotype): set the genotype of the specified index.
- getAC(): get the allele count.
- getAC(int index): get the allele count for the specified allele index.
- getAF(): get the allele frequency.
- getAF(int index): get the allele frequency for the specified allele index.
- getAN(): get the allele number.
- clear(): clear the array, i.e., set all genotypes to
.|.. - toUnPhased(): convert to unphased genotype.
- toPhased(): convert to phased genotype.
- subGenotypes(int[] indexes): get the subgenotype array of the specified indexes.
subGenotypes(BaseArray<Integer> indexes): get the subgenotype array of the specified indexes.- asUnmodifiable(): convert to unmodifiable genotype sequence.
- encode(): encode genotype array.
Create Genotype Array
To create a modifiable genotype array use the edu.sysu.pmglab.gbc.variant.genotype.Genotypes class, the constructor method of this class requires setting the genotype status (phased or unphased) and the number of genotypes. For example:
// Create an unphased genotype array
IGenotypes genotypes = new Genotypes(false, 4);
// Set genotype for each subject
genotypes.setGenotype(0, Genotype.of(0, 1));
genotypes.setGenotype(1, Genotype.of(0, 2));
genotypes.setGenotype(2, Genotype.of(1, 2));
genotypes.setGenotype(3, Genotype.of(2, 3));
The modifiable genotype array allows to modify the genotypes inplace.
Encode Genotype Array
Object coding is termed "serialization" in Java, which means that the object information is stored using a byte stream, and each property, state and method of the object can be restored by "deserialization". IGenotypes automatically selects one of the following five schemes to encode itself:
- NBEG: i.e. number of genotypes is 0
- EBEG: enumerated byte-encodes of genotype, used to store genotypes when a genotype accounts for more than 99.9% of the total number of genotypes.
- MBEG: maximized byte-encodes of genotype, used to store genotypes when the number of optional alleles is 2.
- BEG: single byte-encodes of genotype, used to store genotypes when the number of optional alleles is .
- DBEG: double byte-encodes of genotype, used to store genotypes when the number of optional alleles is .
Examples of the use of encoded and decoded genotype array are as follows:
IGenotypes genotypes = new Genotypes(false, 4);
genotypes.setGenotype(0, Genotype.of(0, 1));
genotypes.setGenotype(1, Genotype.of(0, 2));
genotypes.setGenotype(2, Genotype.of(1, 2));
genotypes.setGenotype(3, Genotype.of(2, 3));
// Encode genotype array, the result is [6, 6, 0, 0, 0, 8, 0, 0, 0, 4, 0, 0, 0, 2, 5, 6, 12]
ByteCode encoded = genotypes.encode();
// Decode encoded genotype sequence
IGenotypes decoded = IGenotypes.load(encoded);
// true
System.out.println(genotypes.equals(decoded));
In the genotype array of a large population, the encoded bytecode usually produces a large number of periodic sequences, which can be further processed using conventional compression methods such as ZSTD and GZIP, all of which can be obtained with good compression effects.
Load an Unmodifiable Genotype Array
Genotype arrays are decoded using IGenotypes.load(encoded) and the resulting object is an unmodifiable cache genotype array object CacheGenotypes.
The cache genotype array stores the original coding sequence, which is returned directly when the coding method encode() is called again. When reading genotypes, the cache genotype sequence uses a fast addressing method to decode specific genotypes in real time (instead of decoding all genotypes), thus achieving low memory loading and high-speed extraction of local genotypes.
If the genotype needs to be modified (e.g., converted to unphased, modified specified individual genotypes), the asModifiable method needs to be called to convert to a modifiable genotype array object (note that a new object is returned).