拆分 GTB
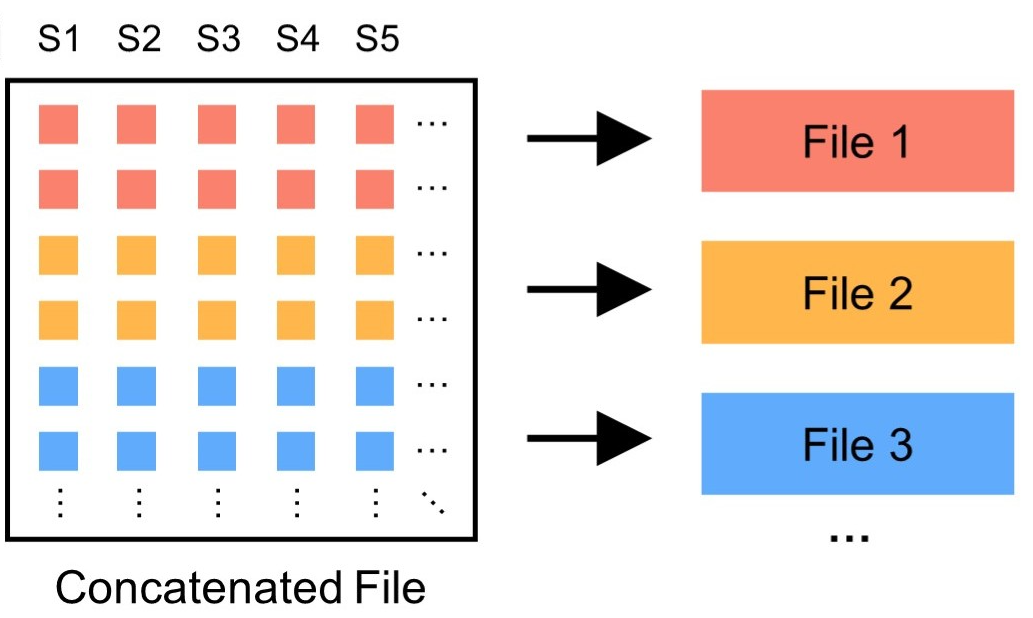
GTB 是一种灵活的可容纳多染色体、万亿数量级位点的储存格式,由于单个文件足以承载大样本全基因组数据,通常情况下无须进行文件拆分。但当单个文件的规模相当庞大时 (通常为大样本量或全基因组位点),切分 GTB 文件为多个子文件有助于进行程序调试和文件传输。使用以下指令对 GTB 文件进行拆分:
java -jar gbc.jar split <input> [output] [options]
拆分后的子文件可以通过 GTBConcat 指令进行重新连接。
[!NOTE|label:示例程序|style:callout]
使用 GBC 为示例文件
https://pmglab.top/gbc/download/rare.disease.hg19.gtb按照变异位点的染色体编号拆分为多个子文件:# 在终端直接运行 java -jar gbc.jar split https://pmglab.top/gbc/download/rare.disease.hg19.gtb # 使用 docker 运行 docker run -v `pwd`:`pwd` -w `pwd` --rm -it -m 4g gbc \ split https://pmglab.top/gbc/download/rare.disease.hg19.gtb
程序参数
语法: split <input> [outputDir] [options]
Java-API: edu.sysu.pmglab.gbc.toolkit.GTBSplitter
关于: 拆分 GTB 文件为多个子文件 (按照染色体或变异位点的索引范围).
参数:
--chromosome 指定染色体标签文件.
格式: --chromosome <file>
--threads,-t 设置并行线程数.
默认值: 4
格式: --threads <int>
--by 按照染色体或变异位点数量切割 GTB 文件为多个子文件.
默认值: chromosome
格式: '--by chromosome [tag],[tag],...' or '--by variant [int]'
子集选择参数:
--pos,-p 提取指定坐标的变异位点.
格式: --pos-index <chr>:<pos>,<pos>,... ... (>= 1)
--pos-range,-pr 提取指定坐标范围的变异位点.
格式: --pos-range <chr>:<minPos>-<maxPos>,... (>= 1)
--index-range,-ir 按照索引提取变异位点.
格式: --index-range <minIndex>-<maxIndex> (>= 0)
--allele-num 排除可变等位基因数不在 [minAlleNum, maxAlleNum] 范围的变异位点.
默认值: 0-15
格式: --allele-num <minAlleleNum>-<maxAlleleNum> (0 ~ 255)
--seq-ac 排除替代等位基因计数 (AC) 不在 [minAc, maxAc] 范围内的变异位点.
格式: --seq-ac <minAc>-<maxAc> (>= 0)
--seq-af 排除替代等位基因频率 (AF) 不在 [minAf, maxAf] 范围内的变异位点.
格式: --seq-af <minAf>-<maxAf> (0.0 ~ 1.0)
--seq-an 排除非缺失等位基因数 (AN) 不在 [minAn, maxAn] 范围内的变异位点.
默认值: 1-
格式: --seq-an <minAn>-<maxAn> (>= 0)
--field-condition 根据指定的补充字段的值提取变异位点. 对于数值字段, 'condition' 格式为 'minValue-maxValue'; 对于其他格式, 'condition'是由 ',' 分隔的多个可选值.
格式: --field-condition <field>=<condition> <field>=<condition> ...
编辑元信息参数:
--add-meta 添加元信息到输出文件.
格式: --add-meta <key>=<value> <key>=<value> ...
--rm-meta 移除所有元信息.
API 工具
对 GTB 文件进行拆分的 API 工具是 edu.sysu.pmglab.gbc.GTBSplitter,使用示例如下:
GTBSplitter.of("https://pmglab.top/gbc/download/rare.disease.hg19.gtb")
.splitByChromosome(null);