基本数据对象
概述
GBC 围绕人类基因组的常见结构设计了相应的 API 对象,数据模型类似于 VCF 文件规范。与 GA4GH 协议 不同的是,GBC 仅要求变异位点对象 Varinat 具有强制的坐标字段,而其他的字段使用键值对的形式储存字段(键的类型是 Object 类型)作为补充字段信息(即属性)。这种方式有助于以标准化的方式共享基因组学数据,节省非必要字段所需的固定内存开销,以及广泛兼容不同的 API 实现。以下是最常用的 API 列表:
- edu.sysu.pmglab.gbc.variant.Chromosome:染色体对象
- edu.sysu.pmglab.gbc.variant.Strand:链方向枚举类,具有 2 个枚举实例:FWD (正链)和 REV(反链)。变异位点对象的链方向强制为 FWD。反链上的变异位点应使用染色体长度调整为 FWD
- edu.sysu.pmglab.gbc.variant.PositionType:坐标系统类型枚举类,具有 2 个枚举实例:ONE_BASED(1-based)和 ZERO_BASED(0-based)。变异位点的坐标系统类型强制为 1-based
- edu.sysu.pmglab.gbc.variant.RefGenomeVersion:参考基因组版本枚举类,具有 4 个枚举实例:hg18、hg19、hg38 和 unknown。非人类参考基因组只能使用 unknown
- edu.sysu.pmglab.gbc.variant.Variant:标准变异位点对象
- edu.sysu.pmglab.gbc.variant.Variants:具有相同坐标的多个变异位点对象
- edu.sysu.pmglab.gbc.variant.genotype.Genotype:基因型对象
- edu.sysu.pmglab.gbc.variant.genotype.IGenotypes:基因型序列对象
下面是创建具有多个可选等位基因、基因型阵列和自定义属性的变异位点的示例:
// 创建一个坐标为 chr1:100 的变异位点, 具有 AT、A、C、TT 四种可变等位基因形式
Variant variant = new Variant("chr1", 100, "AT", "A", "C", "TT");
// 创建一个基因型序列, 基因型为 UnPhased, 受试者数为 4
IGenotypes genotypes = new Genotypes(false, 4);
// 设置每个个体的基因型
genotypes.setGenotype(0, Genotype.of(0, 1));
genotypes.setGenotype(1, Genotype.of(0, 2));
genotypes.setGenotype(2, Genotype.of(1, 2));
genotypes.setGenotype(3, Genotype.of(2, 3));
// 设置位点的属性
variant.setProperty(IGenotypes.class, genotypes);
variant.setProperty("isExon", false);
染色体
Chromosome 采用全局静态类设计,用于规范化不同染色体名称指向的同一实体。以下用于获取名称为 “chr1” 的染色体对象:
Chromosome chromosome1 = Chromosome.get("chr1");
它与 GenBank 中的命名 CM000663.2 的染色体关联到同一个染色体对象:
// 结果为 true
chromosome1.equals(Chromosome.get("CM000663.2"))
有关染色体标签的定义、染色体级别属性和说明请参阅:染色体标签声明。
由于不同 VCF 文件、不同数据库文件使用的染色体标签系统可能不同(例如 dbSNP 中的染色体采用 NCBI Reference Sequence 命名),使用常规的字符串格式进行判断无法映射(或需要复杂的设计)到同一实体,我们建议所有的染色体对象都使用 Chromosome.get(Object object) 映射为统一的对象。
若需要为染色体对象赋予其他替代名称或设置不同的物种染色体,参考下例:
// 清除原有的染色体定义
Chromosome.clear();
// 新建染色体标签
Chromosome chromosome1 = Chromosome.addChromosome(new Chromosome("1", "chr1"));
// 设置染色体属性
chromosome1.setProperty("length", 249250621);
// 获取染色体属性
chromosome1.getProperty("length");
坐标系统
基因组学中的坐标系统 PositionType 有两种:0-based 和 1-based。其中,0-based 坐标系统是指从 0 开始计数,而 1-based 坐标系统是指从 1 开始计数。在基因组学中,常用的文件格式 SAM、GFF/GTF、VCF、WIG、Axt、BLAST 比对结果等,这些文件格式通常采用 1-based 坐标系统。而 BAM、PSL 以及 BED 等文件格式则采用 0-based 坐标系统。
对于一段序列 ATGC 而言,碱基 T 在 0-based 下的坐标是 1,而在 1-based 下的坐标是 2。对于子序列 TG,在 0-based 下的坐标是 [1, 3),而在 1-based 下的坐标是 [2, 3]。
参考基因组版本
人类参考基因组版本是由参考基因组联盟(Genome Reference Consorium)发布的,目前广泛使用的版本是 GRCh37 和 GRCh38 (hg19 和 hg38 是对应的 UCSC 浏览器版本)。两个版本之间的区别主要在于:1)hg38 比 hg19多了一些测序位点,导致整个基因组的位置都发生了偏移;2)hg38 相对于 hg19 是一个巨大进步,改变了之前的一些测序错误,组装错误。
通常情况下 GBC 并不要求指定参考基因组版本 RefGenomeVersion,仅当用户使用公共数据库进行功能注释时参考基因组版本是必须的。GBC 的多个功能(如构建和编辑 GTB 文件)都提供参考基因组版本转换的功能。
API 方法 edu.sysu.pmglab.gbc.annotation.impl.liftOver.LiftOver 用于支持参考基因组版本转换,LiftOver 接收一个 Chain 文件 作为输入(或使用 edu.sysu.pmglab.gbc.annotation.impl.liftOver.LiftOver.Chain 类构造区间映射关系)。GBC 默认提供 hg18, hg19, hg38, unknown 四种参考基因组版本,并内置了 hg18ToHg19, hg18ToHg38, hg19ToHg38, hg38ToHg19 的坐标转换功能。常用的 Chain 文件可以在 UCSC 网站 下载。例如:
- hg19ToHg38:https://hgdownload.soe.ucsc.edu/goldenPath/hg19/liftOver/hg19ToHg38.over.chain.gz
- hg38ToHg19:https://hgdownload.soe.ucsc.edu/goldenPath/hg38/liftOver/hg38ToHg19.over.chain.gz
LiftOver hg19ToHg38LiftOver;
// 使用内置的 hg19ToHg38, 它从 UCSC 网站下载相应的 LiftOver 文件
hg19ToHg38LiftOver = LiftOver.load(RefGenomeVersion.hg19, RefGenomeVersion.hg38);
System.out.println(hg19ToHg38LiftOver.convertCoordinate(Chromosome.get("chr8"), 141310715));
// 使用 UCSC 文件地址, 地址可以是本地文件路径或 http/https 文件路径
hg19ToHg38LiftOver = LiftOver.load("https://hgdownload.soe.ucsc.edu/goldenPath/hg19/liftOver/hg19ToHg38.over.chain.gz");
System.out.println(hg19ToHg38LiftOver.convertCoordinate(Chromosome.get("chr8"), 141310715));
LiftOver 具有五种转换方法:
- convertCoordinate(Chromosome chromosome, int position):对指定坐标(1-based)进行版本转换,返回坐标在正链上的映射值
- 当源坐标无法映射到目标参考基因组版本时,它返回 null
- 返回值是
Entry<Chromosome, Integer>对象,使用 getKey() 获得染色体,使用 getValue() 获得坐标值
- convertCoordinate(Chromosome chromosome, int position, PositionType positionType):对指定坐标进行版本转换,返回坐标在正链上的映射值
- 当源坐标无法映射到目标参考基因组版本时,它返回 null
- 返回值是
Entry<Chromosome, Integer>对象,使用 getKey() 获得染色体,使用 getValue() 获得坐标值
- convertCoordinate(Variant variant):对变异位点进行版本转换
- 当源坐标无法映射到目标参考基因组版本时,它返回 null
- 返回值是一个具有新坐标的 Variant 对象,当返回的 Variant 对象与输入 Variant 对象的坐标相同时,将返回输入值本身
- convertInterval(Chromosome chromosome,
Interval<Integer>positions):对指定坐标区间(0-based)进行版本转换,返回坐标在正链上的映射值- 当源坐标起点或终点无法映射到目标参考基因组版本、映射前后的区间长度不一致、映射后的起点和终点坐标不在同一染色体上时,它返回 null
- 返回值是
Entry<Chromosome, Interval<Integer>>对象,使用 getKey() 获得染色体,使用 getValue() 获得区间
- convertInterval(Chromosome chromosome,
Interval<Integer>positions, PositionType positionType):对指定坐标区间进行版本转换,返回坐标在正链上的映射值- 当源坐标起点或终点无法映射到目标参考基因组版本、映射后的区间长度不一致、映射后不在同一染色体上时,它返回 null
- 返回值是
Entry<Chromosome, Interval<Integer>>对象,使用 getKey() 获得染色体,使用 getValue() 获得区间
当您在开发一个支持 LiftOver 的功能时(例如变异功能注释),我们强烈建议将 “不进行参考基因组版本转换” 的值为 LiftOver.EMPTY,而不是 null(这样可以保证方法的上下文统一,避免频繁判断 LiftOver 是否为 null)。LiftOver.EMPTY 是 LiftOver 的静态实例,它将所有的输入原封不动地返回。
标准变异位点
单位点对象
标准变异位点对象 Variant 用于表示一个坐标 (chromosome, position) 上的变异信息,它包含两个强制的、不可变的成员变量:
- chromosome:染色体对象,不允许为 null
- position:坐标值,要求为正链上的 1-based 坐标,值为正整数
以及可选的成员变量:
- alleles:可选等位基因列表,第一个可选等位基因对应为 VCF 文件规范 中的 REF,其余可选等位基因对应为 ALT
- 通过 addAlleles(String... alleles) 或 addAlleles(ByteCode... alleles) 添加等位基因
- 通过 alleleOfIndex(int index) 获取指定索引的等位基因 (索引 0 对应的等位基因为参考等位基因),超过索引范围时返回 null
- 通过 indexOfAllele(String allele) 或 indexOfAllele(ByteCode allele) 获取指定等位基因对应的索引,指定等位基因不存在时返回 -1
- 通过 isStandardAllele(String allele) 或 isStandardAllele(ByteCode allele) 判断指定等位基因是否为标准等位基因 (即所有的碱基都是 A、T、C、G)
- 通过 isValidAllele(String allele) 或 isValidAllele(ByteCode allele) 判断指定等位基因是否为合法等位基因 (即不能为
,;和不可见字符) - 通过 getAlleles() 获取该位点的所有可选等位基因
- 通过 getAlleleNum() 获取该位点的可选等位基因个数
- 通过 clearAlleles() 清空该位点的等位基因
此外,使用 setProperty(Object key, Object value) 设置位点属性值,使用 getProperty(Object key) 获取属性值。例如,GBC 中的标准基因型序列对象使用 getProperty(IGenotypes.class) 获得。
除了以上成员变量及方法外,变异位点还提供一个标准化位点方法 normalized(),用于将一个多等位基因位点转为多个二等位基因位点,并校正后向冗余碱基。单位点对象进行 normalized() 时的返回值是一个多位点对象,可用于方便地表示每一种突变形式的功能注释。以下是一个多等位基因位点进行标准化的示例:
#CHROM POS REF ALT S1 S2 S3 S4 isExon
1 100 AT A,C,TT 0/1 0/2 1/2 2/3 false
被标准化为:
#CHROM POS REF ALT S1 S2 S3 S4 isExon
1 100 AT A 0/1 0/0 0/1 0/0 false
1 100 AT C 0/0 0/1 0/1 0/1 false
1 100 A T 0/0 0/0 0/0 0/1 false
请注意,在这种情况下基因型编号 0 代表 “非 ALT 基因型”,在确定个体基因型时需要扫描所有相同坐标的位点的基因型。单位点对象的属性值被设置为多位点对象的属性,如需为子位点设置同名属性,通过 get(int index) 方法获得特定索引的子变异位点进行设置(优先级:同名子位点属性 > 同名主位点属性)。
[!TIP|label:如何兼容其他 API 规范中的变异位点对象|style:callout]
将其他的 API 实现作为变异位点的属性:
variant.setProperty(org.cobi.kggsee.entity.Variant.class, kggseeVariant);
多位点对象
多位点对象 Variants 用于表示多个具有相同坐标 (chromosome, position) 上的变异位点,它同样包含强制字段 chromosome 和 position。使用 add(Variant variant) 和 addAll(Iterable<Variant> variants) 向该对象中添加变异位点。get(int index) 方法用于获取该对象中的索引为 index 的子变异位点,而 size() 方法用于获取该对象包含的子位点的个数。
与单位点对象相同,使用 setProperty(Object key, Object value) 设置位点属性值,使用 getProperty(Object key) 获取属性值。此时,这些设置在多位点对象上的属性表示所有子位点对象的共同属性。
基因型
基因型类 Genotype 用于标准化单倍体和二倍体物种的基因型,它至多表示包含 255 种可变的等位基因形式的基因型(即基因型 a|b 中每一个基因型值都不超过 254)。
在 GBC 中使用单例对象表示基因型,这使得所有的基因型 a|b 都定位到具有相同内存地址的基因型对象。缺失基因型使用 Genotype.of(-1, -1) 或 Genotype.MISS_GENOTYPE 获得基因型对象;二倍体基因型 a|b 或 a/b 使用 Genotype.of(a, b) 获得基因型对象;单倍体基因型 a 使用 Genotype.of(a, a) 获得基因型对象。
该类的实例对象提供以下方法:
- reverse():将基因型
a|b翻转为b|a,并返回指向b|a的基因型实例 - toUnPhased():将基因型
a|b转为 UnPhased 的形式(即左侧基因型 右侧基因型),并返回基因型实例 - getLeftGenotype():获得基因型
a|b的左侧基因型 - getRightGenotype():获得基因型
a|b的右侧基因型 - isMissingGenotype():该基因型是否为缺失基因型
- isHomozygous():该基因型是否为纯合子
- isHeterozygous():该基因型是否为杂合子
基因型编码
每个基因型都被映射(哈希值)为 0-65025 范围内的唯一整数。对于缺失基因型 .|.,它被映射为 0。对于一个非缺失基因型 a|b,它被映射为
完整的基因型映射表请参阅 BEG.xlsx。基因型编码包括单字节、双字节两种形式。当每一侧的基因型值不超过 14 时,基因型的编码值为其单字节哈希值(此时,哈希值不超过 211)。当至少一侧基因型值超过 14 时,基因型的编码值为其哈希值的双字节编码形式。例如,基因型 20|30 的哈希值为 921,其字节编码值为:[-103, 3]。
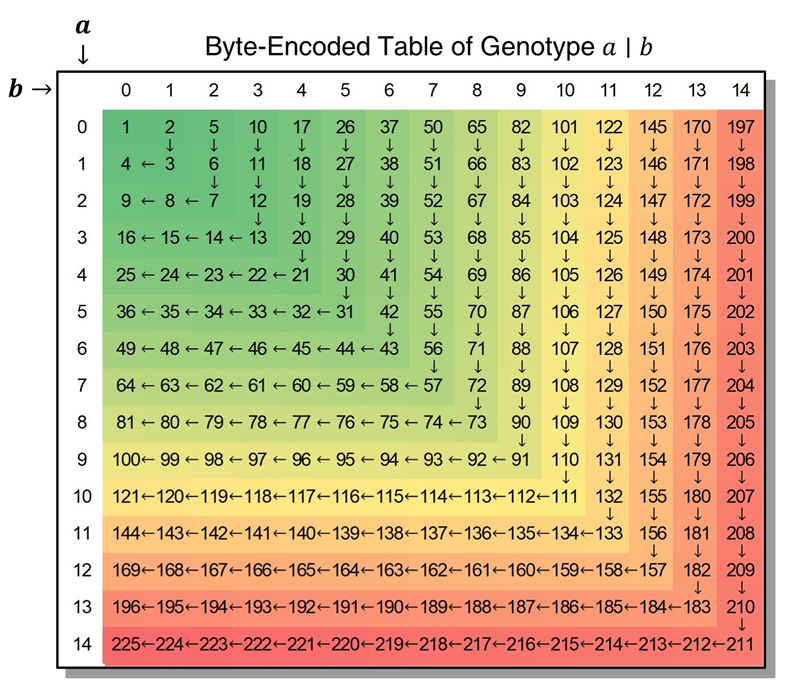
基因型序列
基因型序列类 IGenotypes 用于以尽可能少的内存开销储存多个样本在同一坐标上的基因型。当一个变异位点包含受试者基因型时,这些基因型储存在变异位点对象的 IGenotype.class 属性上。
基因型序列包括以下几个常用的方法:
- getGenotype(int index):获取指定索引的基因型
- getLeftGenotype(int index):获取指定索引的左侧基因型
- getRightGenotype(int index):获取指定索引的右侧基因型
- setGenotypes(int index, Genotype genotype):设置指定索引的基因型
- getAC():获取等位基因计数 (allele count)
- getAC(int index):获取指定等位基因索引的等位基因计数
- getAF():获取等位基因频率 (allele frequency)
- getAF(int index):获取指定等位基因索引的等位基因频率
- getAN():获取等位基因总数 (allele number)
- clear():清空基因型序列
- toUnPhased():转为 UnPhased 基因型
- toPhased():转为 Phased 基因型
- toBiallelic():转为二等位基因位点(所有非
0和非空的基因型都被设置为1) - subGenotypes(int[] indexes):获取指定索引的子基因型序列
subGenotypes(BaseArray<Integer> indexes):获取指定索引的子基因型序列- asUnmodifiable():转为不可变的基因型序列
- encode():基因型序列编码
创建可变基因型序列
创建一个可变基因型序列使用 edu.sysu.pmglab.gbc.variant.genotype.Genotypes 类,该类的构造器方法要求设置基因型的向型和基因型个数。例如:
// 创建一个基因型序列, 基因型为 UnPhased, 受试者数为 4
IGenotypes genotypes = new Genotypes(false, 4);
// 设置每个个体的基因型
genotypes.setGenotype(0, Genotype.of(0, 1));
genotypes.setGenotype(1, Genotype.of(0, 2));
genotypes.setGenotype(2, Genotype.of(1, 2));
genotypes.setGenotype(3, Genotype.of(2, 3));
可变基因型序列允许直接原位修改基因型的值,通常用于 “创建” 时使用。
编码基因型序列
对象编码在 Java 中的术语为 “对象序列化”,即使用字节流的形式储存对象信息,并可通过 “反序列化” 还原对象的各个属性、状态和方法。IGenotypes 根据基因型的可变等位基因数及基因型种类,自动选择以下五种类型之一进行基因型序列的编码:
- NBEG 编码:无基因型占位编码(即基因型个数为 0)
- EBEG 编码:枚举基因型字节编码,当某种基因型占比超过 99.9% 时,使用此编码储存基因型
- MBEG 编码:最简基因型字节编码,当可变等位基因数为 2 时,使用此编码储存基因型
- BEG 编码:单字节基因型编码,当可变等位基因数 时,使用此编码储存基因型
- DBEG 编码:双字节基因型编码,当可变等位基因数 时,使用此编码储存基因型
[!NOTE|label:基因型序列编码细节|style:callout]
NBEG 编码使用一个字节表示编码结果,0 代表无向型 NBEG 序列,1 代表有向型 NBEG 序列。其他编码则为“前缀码 + 序列编码”的形式。
前缀码的形式为:
[向型码 + (类型码 << 1)][AC][AN][Genotype Num]
- 向型码:向型为 Phased 时,向型码为 1,否则为 0
- 类型码:按照以上顺序依次为 0, 1, 2, ...
- AC:等位基因计数值,编码为 4 字节
- AC:等位基因总数,编码为 4 字节
- Genotype Num:基因型个数,编码为 4 字节
此后,使用以下策略进行基因型数据的编码
- EBEG 编码:
[主要基因型][次要基因型1:个数,索引1,索引2,...][次要基因型2:个数,索引1,索引2,...],主要基因型与次要基因型都使用双字节形式编码,个数、索引都各自被编码为 4 字节长度- MBEG 编码:
[组合基因型1, 1字节][组合基因型2, 1字节]...- BEG 编码:
[基因型1, 1字节][基因型2, 1字节]...- DBEG 编码:
[基因型1, 2字节][基因型2, 2字节]...有关 MBEG 编码的实施细节,请参阅 BEG.xlsx。
编码与解码的使用示例如下:
IGenotypes genotypes = new Genotypes(false, 4);
genotypes.setGenotype(0, Genotype.of(0, 1));
genotypes.setGenotype(1, Genotype.of(0, 2));
genotypes.setGenotype(2, Genotype.of(1, 2));
genotypes.setGenotype(3, Genotype.of(2, 3));
// 编码基因型序列, 结果 [6, 6, 0, 0, 0, 8, 0, 0, 0, 4, 0, 0, 0, 2, 5, 6, 12]
ByteCode encoded = genotypes.encode();
// 解码基因型序列
IGenotypes decoded = IGenotypes.load(encoded);
// 判断两个基因型序列是否相等, 返回值 true
System.out.println(genotypes.equals(decoded));
在大规模人群的基因型序列中,编码后的字节码通常会产生大量的周期性序列,使用常规的 ZSTD、GZIP 等压缩方法进一步处理,都能获得良好的压缩效果。
加载不可变基因型序列
基因型序列使用 IGenotypes.load(encoded) 解码后产生的对象不再是可变基因型序列对象 Genotypes,而是不可变的缓存型基因型序列对象 CacheGenotypes。
缓存型基因型序列储存着原始编码信息,当再次调用编码方法 encode() 时,直接返回该编码信息。读取基因型时,缓存型基因型序列使用快速寻址方法实时进行特定基因型的解码(而不是解码全部的基因型),以此实现低内存载入、高速提取局部基因型的特点。
若需要对基因型发生修改(例如转为 unphased、修改个体基因型)时,需要调用 asModifiable 方法转换为可变基因型序列对象(注意,返回时是一个新对象)。
[!NOTE|label:基因型序列解码细节|style:callout]
- 若基因型序列编码长度为 1,则 0 代表无向型 NBEG,1 代表有向型 NBEG
- 根据编码序列第一个字节
(bytecode.byteAt(0) >> 1) & 0xFF获取对应的类型码,根据bytecode.byteAt(0) & 0x01获取对应的向型- 根据编码序列的第 2~5 字节、6~9 字节、10~13 字节分别还原 AC、AN、Genotype Num
- 每一种类型使用独立的解码策略
- EBEG 编码:将主基因型设置为 majorGenotype,其余基因型还原为
Map<Integer, Genotype> cache的形式;查询的索引不包含在cache中时返回 majorGenotype,否则返回对应的基因型对象- MBEG 编码:对于查询的基因型索引
index
- UnPhased MBEG 编码:使用
MBEGDecoder.UN_PHASED.decode(bytecode.byteAt(13 + (index >> 2)), index & 3)获取基因型对象- Phased MBEG 编码:使用
MBEGDecoder.PHASED.decode(bytecode.byteAt(13 + (index / 3)), index % 3)获取基因型对象- BEG 编码:对于查询的基因型索引
index,使用Genotype.of(13 + index)获取基因型对象- DBEG 编码:对于查询的基因型索引
index,使用Genotype.of(decodeShort(bytecode.byteAt(13 + (index << 1)), bytecode.byteAt(14 + (index << 1))))获取基因型对象